



El equipo científico liderado por el catedrático de Biología Celular de la Universidad Pablo de Olavide Carlos Santos Ocaña, participa en un estudio internacional que demuestra cómo la deficiencia de CoQ10 asociada a problemas de ataxia y alteraciones del neurodesarrollo en un grupo de pacientes que provienen de tres familias no relacionadas, se deben a mutaciones en el gen GEMIN5. Este gen codifica una proteína que es componente del complejo de supervivencia de la motoneurona (SMN) cuyas mutaciones producen la Atrofia Muscular Espinal y que hasta ahora no había sido relacionado con disfunción mitocondrial y deficiencia de CoQ10.
Esta investigación abre una nueva vía terapéutica que permitirá a otros pacientes con mutaciones en GEMIN5, que no se habían relacionado con la deficiencia de CoQ10, disponer de una terapia efectiva que hasta el momento no existía. Además, abre una nueva línea de trabajo que relaciona el desarrollo del sistema nervioso con la función mitocondrial.
Los resultados de este estudio han sido publicados en European Journal of Human Genetics fruto de una colaboración entre el grupo del Dr. Artuch en el Hospital San Joan de Deu en Barcelona y del Dr. Pandei en el Hospital Pediátrico de Pittsburg con el grupo de investigación de la Universidad Pablo de Olavide, que desarrolla sus estudios en el Centro Andaluz de Biología del Desarrollo (centro mixto de la UPO, CSIC y Junta de Andalucía) y que forma parte del Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). La colaboración con el Dr. Artuch forma parte de un proyecto de investigación financiado por el Instituto de Salud Carlos III que está orientado al diagnóstico, esclarecimiento de los mecanismos de la enfermedad y la terapia de enfermedades mitocondriales.
La investigación, paso a paso
El trabajo se inició en 2004 analizando un caso de posible deficiencia de CoQ10 asociada a ataxia cerebelosa en una paciente de 12 años. La ataxia cerebelosa produce problemas para el control de los movimientos musculares voluntarios. “A pesar de los esfuerzos no se pudo identificar la causa genética de la enfermedad, pero la rápida administración de CoQ10 produjo una clara mejoría de los síntomas, que ha permitido llevar una vida casi normal a la paciente durante los últimos 20 años”, explica Carlos Santos Ocaña.
En el año 2020, la aplicación de nuevas técnicas de secuenciación de ADN permitió identificar a una mutación en el gen GEMIN5 como la causa genética de la enfermedad. La búsqueda de nuevos casos en España permitió encontrar dos nuevas familias, no relacionadas con la anterior, que también mostraron deficiencia de CoQ10 asociado a ataxia cerebelosa y alteraciones del neurodesarrollo, y que han podido ser tratados con CoQ10.
“El gen GEMIN5 es necesario para producir una proteína que es componente del complejo de supervivencia de la motoneurona (SMN), cuyas mutaciones producen la Atrofia Muscular Espinal y que hasta ahora no había sido relacionado con disfunción mitocondrial y deficiencia de CoQ10” explica Santos Ocaña, quien resalta la esencial colaboración con el Dr. Pandei en Pittsburg al haber coordinado recientemente un estudio sobre 32 pacientes con alteraciones del neurodesarrollo que portaban mutaciones del gen GEMIN5, cuya enfermedad no se había asociado a una disfunción mitocondrial ni a una deficiencia de CoQ10.
Doble papel de GEMIN5
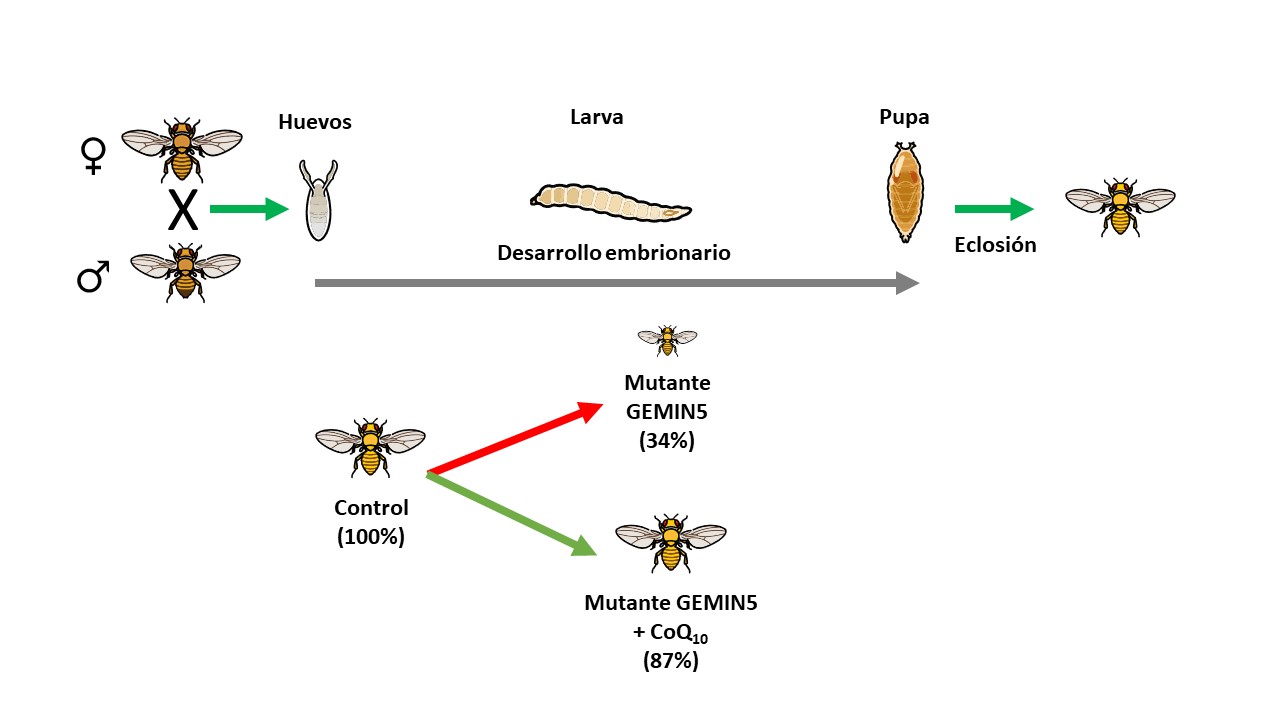
 Uno de los resultados de este estudio ha sido mostrar cómo las mutaciones en GEMIN5 generan una disfunción mitocondrial en las células de pacientes, que puede ser revertida total o parcialmente mediante la administración de CoQ10.
Uno de los resultados de este estudio ha sido mostrar cómo las mutaciones en GEMIN5 generan una disfunción mitocondrial en las células de pacientes, que puede ser revertida total o parcialmente mediante la administración de CoQ10.
Además, el equipo investigador ha demostrado que la mutación de GEMIN5 en el modelo de Drosophila melanogaster (la mosca de la fruta) genera una grave alteración neurológica, que afecta al desarrollo larvario, y que se centra en el control de movimientos dificultando el salto o la búsqueda de alimentos. En este modelo experimental la administración de CoQ10 permitió revertir los defectos causados por la mutación en GEMIN5 en el modelo de mosca de la fruta.
“La importancia de este estudio radica en las posibilidades terapéuticas que ofrece. Hasta el momento, los pacientes con mutaciones en GEMIN5 no disponían de un tratamiento efectivo, pero, con la identificación de la causa genética de la enfermedad, el tratamiento con CoQ10 podría permitir mejorar sustancialmente su calidad de vida” explica el investigador de la UPO.
Además de la vía terapéutica, este trabajo abre una nueva línea de trabajo que relaciona claramente el desarrollo del sistema nervioso con la función mitocondrial. La proteína GEMIN5 tiene un papel fundamental en la diferenciación de las neuronas, las células nerviosas y, sobre todo, las motoneuronas. Estas células son las encargadas transmitir al músculo la señal proveniente del cerebro para iniciar y regular la contracción muscular. Ello explica los síntomas detectados en los pacientes.
Sin embargo, GEMIN5 también parece mostrar un papel adicional regulando la generación de nuevas mitocondrias, los responsables de la generación de energía en la célula y que depende de la producción de unos niveles adecuados de CoQ10.
Este doble papel de GEMIN5 pivota en su capacidad para regular los niveles de CoQ10, que pueden incrementarse mediante el uso del ubiquinol o CoQ10 como terapia efectiva. “En este sentido nuestro grupo de investigación, junto con el grupo del Dr. Artuch, somos responsables de la aprobación en 2021 por parte de la Agencia Europea del Medicamento del ubiquinol como medicamento huérfano para las ataxias generadas por deficiencia de CoQ10, y participamos durante el año 2023 en un estudio clínico dirigido por la Dra. Angels García Cazorla del Hospital San Joan de Deu denominado Eficacia y tolerabilidad de la coenzima Q (Ubiquinol) en pacientes con trastornos mitocondriales y ataxias cerebelosas financiado por el Ministerio de Ciencia e Innovación”, concluye Carlos Santos Ocaña.
Referencia:
Cascajo-Almenara, M.V., Juliá-Palacios, N., Urreizti, R. et al. Mutations of GEMIN5 are associated with coenzyme Q10 deficiency: long-term follow-up after treatment. European Journal of Human Genetics (2024). https://doi.org/10.1038/s41431-023-01526-2











