


Las enfermedades mitocondriales representan uno de los grupos más comunes de enfermedades genéticas. Con una prevalencia superior a 1 en 5000 adultos, la mayoría de ellas todavía carecen de un tratamiento eficaz. Las terapias actuales son puramente paliativas y, en la mayoría de los casos, insuficientes, de ahí que la comunidad científica deba desarrollar nuevos enfoques para compensar y, si es posible, revertir la disfunción mitocondrial.
En esta línea trabaja el equipo científico de la Universidad Pablo de Olavide, dirigido por el profesor José Antonio Sánchez Alcázar, que ha publicado recientemente un estudio que demuestra cómo las alteraciones patológicas en la mutación mitocondrial GFM1 pueden ser corregidas por activadores de la UPRmt, dando un paso adelante en la estrategia terapéutica para las enfermedades mitocondriales.

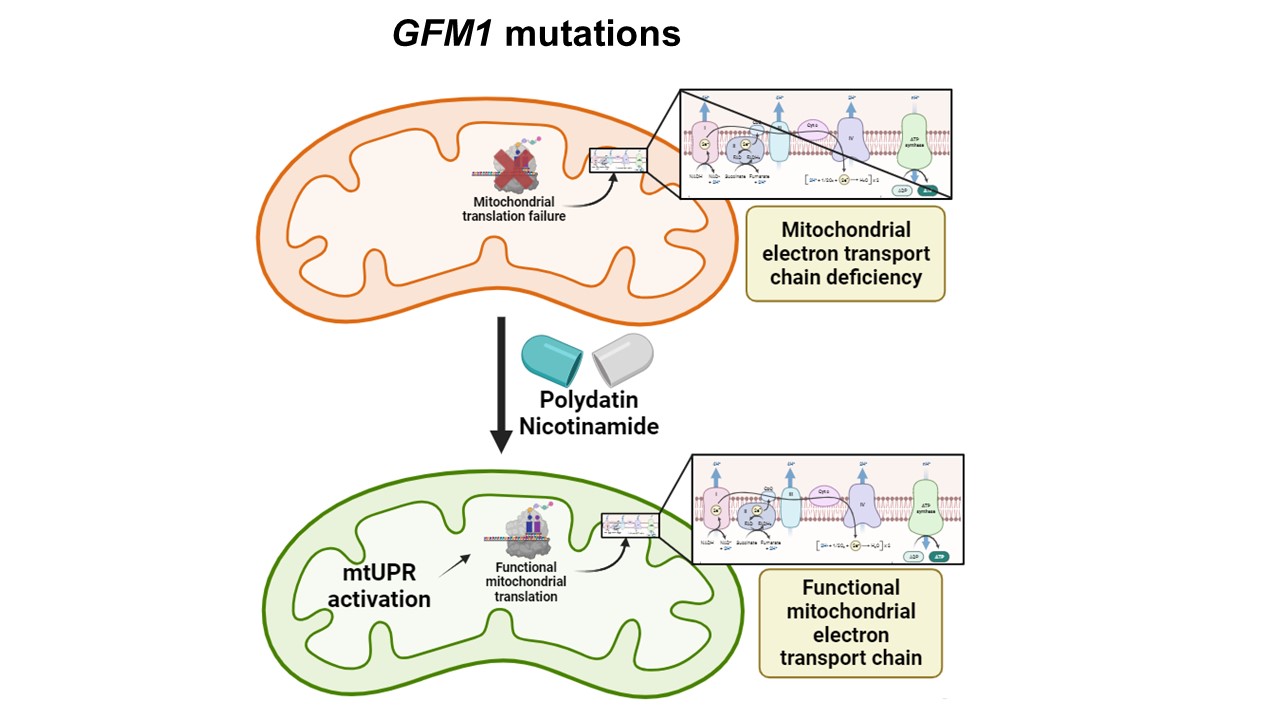
En el estudio, el grupo del doctor Sánchez Alcázar abordó el problema utilizando como modelo biológico los fibroblastos de tres pacientes portadores de mutaciones en el gen GFM1, que está implicado en la síntesis de proteínas mitocondriales. “La mutación GFM1 causa deficiencias de la cadena respiratoria y cuadros clínicos muy graves. Los fibroblastos GFM1 mutantes no pueden sobrevivir en un medio con galactosa que fuerza a las células a depender exclusivamente de la función mitocondrial”, explican los investigadores. “Sin embargo, el tratamiento con polidatin y nicotinamida permitió la supervivencia de los fibroblastos GFM1 mutantes en este medio”, añaden.
Las mutaciones de GFM1 bloquean la síntesis de las proteínas mitocondriales comprometiendo la producción energética mitocondrial. El tratamiento con polidatin y nicotinamida activa la UPRmt y mejora la función de la proteína mutante y la bioenergética mitocondrial.
Con este trabajo, los investigadores e investigadoras de la UPO demuestran que la combinación de polidatin y nicotinamida activa la respuesta a proteínas mal plegadas mitocondrial (UPRmt), una vía compensatoria que regula el correcto funcionamiento mitocondrial. Además, comprobaron que la activación de UPRmt mejora la bioenergética celular de las células mutantes y restaura parcialmente la expresión de las proteínas mitocondriales. Los resultados fueron confirmados en neuronas inducidas obtenidas por reprogramación directa de los fibroblastos del paciente.
“El estudio propone que la activación de los mecanismos compensatorios celulares como la UPRmt es una estrategia terapéutica prometedora para las enfermedades mitocondriales”, afirman.
La aplicación del cribado personalizado con activadores de la UPRmt abre de esta manera una nueva ventana de posibilidades para el tratamiento de las patologías genéticas mitocondriales. Las mutaciones estudiadas por este grupo de investigación afectan tanto a genes localizados en el ADN nuclear (COQ7, COX15, NDUFAF6, NDUFS1 y NDUFS4) como en el ADN mitocondrial (ND3).
Con el objetivo de trasladar los resultados de la investigación a los pacientes, el grupo del Dr. Sánchez Alcázar colabora estrechamente con el equipo del Dr. Andrés Rodríguez Sacristán de la Unidad de Neuropediatría del Servicio de Pediatría del Hospital Universitario Virgen Macarena de Sevilla, para la realización de un ensayo clínico en las enfermedades mitocondriales aplicando la aproximación de la medicina de precisión personalizada.
Plataforma MITOCURE: Medicina de precisión personalizada en las enfermedades mitocondriales
Las enfermedades mitocondriales abarcan un amplio espectro de trastornos musculares y neurodegenerativos, crónicos y progresivos, causadas por mutaciones en el ADN nuclear (nDNA) o mitocondrial (mtDNA), la mayoría de las cuales no tienen tratamiento eficaz. Estas enfermedades tienen una gran heterogeneidad y afectan fundamentalmente a la capacidad de producción energética de las células.
Las terapias farmacológicas actuales se basan fundamentalmente en eliminar los metabolitos tóxicos; intentar circunvalar los bloqueos de la cadena respiratoria; administrar metabolitos y cofactores para mejorar la síntesis de ATP; y prevenir el estrés oxidativo.
Dada la diversidad de mutaciones y las diferentes opciones terapéuticas, la propuesta de MITOCURE defiende que en las enfermedades mitocondriales es obligado una aproximación terapéutica personalizada.
Con esta plataforma los investigadores e investigadoras de la UPO evalúan la efectividad terapéutica de los distintos tratamientos disponibles en los fibroblastos derivados de los pacientes mitocondriales y en las células neuronales generadas por reprogramación directa. Para conseguir este objetivo, estudian los efectos de estos tratamientos sobre las alteraciones fisiopatológicas presentes en los fibroblastos y células neuronales derivadas de los pacientes de una manera personalizada.
En los modelos celulares estudian la proliferación celular y/o muerte celular en medio con galactosa (que fuerza la obtención de energía por la mitocondria), las actividades enzimáticas de la cadena respiratoria mitocondrial, los niveles de expresión de las proteínas mitocondriales, el potencial de membrana mitocondrial, y la activación de la degradación de las mitocondrias y/o la apoptosis.
En la actualidad, la plataforma MITOCURE está realizando medición de precisión personalizada en más de 30 mutaciones que afectan directa o indirectamente a la formación de energía por las mitocondrias.
Este grupo de investigación de la Universidad Pablo de Olavide, que desarrolla su trabajo en el Centro Andaluz de Biología del Desarrollo (centro mixto del CSIC, UPO y Junta de Andalucía), aplica este método de trabajo basado en la medicina de precisión personalizada, además de en el proyecto MITOCURE, en sus diferentes proyectos, como MYOCURE (centrado en las miopatías congénitas), BRAINCURE (centrado en neurodegeneración por acumulación de hierro y enfermedades desmielinizantes), o NEK8 (centrado en mutaciones en el gen NEK8).
Artículo de referencia:
Paula Cilleros-Holgado, David Gómez-Fernández, Rocío Piñero-Pérez, José Manuel Romero Domínguez, Marta Talaverón-Rey, Diana Reche-López, Juan Miguel Suárez-Rivero, Mónica Álvarez-Córdoba, Ana Romero-González, Alejandra López-Cabrera, Marta Castro De Oliveira, Andrés Rodríguez-Sacristán, José Antonio Sánchez-Alcázar. Polydatin and nicotinamide rescue the cellular phenotype of mitochondrial diseases by mitochondrial unfolded protein response (mtUPR) activation. Biomolecules, 2024, 14(5), 598. https://doi.org/10.3390/biom14050598
Referencias del grupo de investigación:
https://sanchezalcazarlab.com/
https://pubmed.ncbi.nlm.nih.gov/?term=sanchez+alcazar&sort=pubdate
https://www.researchgate.net/profile/Jose-Sanchez-Alcazar











